See on Scoop.it – Curiosopernatura
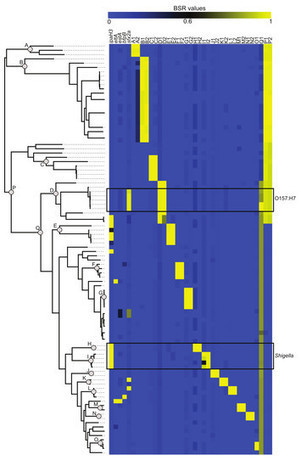
Background. As whole genome sequence data from bacterial isolates becomes cheaper to generate, computational methods are needed to correlate sequence data with biological observations. Here we present the large-scale BLAST score ratio (LS-BSR) pipeline, which rapidly compares the genetic content of hundreds to thousands of bacterial genomes, and returns a matrix that describes the relatedness of all coding sequences (CDSs) in all genomes surveyed. This matrix can be easily parsed in order to identify genetic relationships between bacterial genomes. Although pipelines have been published that group peptides by sequence similarity, no other software performs the rapid, large-scale, full-genome comparative analyses carried out by LS-BSR.Results. To demonstrate the utility of the method, the LS-BSR pipeline was tested on 96 Escherichia coli and Shigella genomes; the pipeline ran in 163 min using 16 processors, which is a greater than 7-fold speedup compared to using a single processor. The BSR values for each CDS, which indicate a relative level of relatedness, were then mapped to each genome on an independent core genome single nucleotide polymorphism (SNP) based phylogeny. Comparisons were then used to identify clade specific CDS markers and validate the LS-BSR pipeline based on molecular markers that delineate between classical E. coli pathogenic variant (pathovar) designations. Scalability tests demonstrated that the LS-BSR pipeline can process 1,000 E. coli genomes in 27–57 h, depending upon the alignment method, using 16 processors.Conclusions. LS-BSR is an open-source, parallel implementation of the BSR algorithm, enabling rapid comparison of the genetic content of large numbers of genomes. The results of the pipeline can be used to identify specific markers between user-defined phylogenetic groups, and to identify the loss and/or acquisition of genetic information between bacterial isolates. Taxa-specific genetic markers can then be translated into clinical diagnostics, or can be used to identify broadly conserved putative therapeutic candidates.
See on peerj.com