See on Scoop.it – Curiosopernatura
Abstract
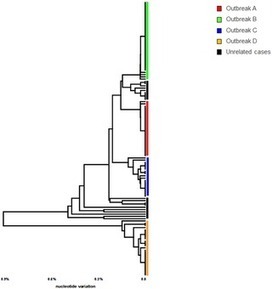
The genetic characterization of hepatitis A virus (HAV) strains is commonly accomplished by sequencing subgenomic regions, such as the VP1/P2B junction. HAV genome is not extensively variable, thus presenting opportunity for sharing sequences of subgenomic regions among genetically unrelated isolates. The degree of misrepresentation of phylogenetic relationships by subgenomic regions is especially important for tracking transmissions. Here, we analyzed whole-genome (WG) sequences of 101 HAV strains identified from 4 major multi-state, food-borne outbreaks of hepatitis A in the Unites States and from 14 non-outbreak-related HAV strains that shared identical VP1/P2B sequences with the outbreak strains. Although HAV strains with an identical VP1/P2B sequence were specific to each outbreak, WG were different, with genetic diversity reaching 0.31% (mean 0.09%). Evaluation of different subgenomic regions did not identify any other section of the HAV genome that could accurately represent phylogenetic relationships observed using WG sequences. The identification of 2–3 dominant HAV strains in 3 out of 4 outbreaks indicates contamination of the implicated food items with a heterogeneous HAV population. However, analysis of intra-host HAV variants from eight patients involved in one outbreak showed that only a single sequence variant established infection in each patient. Four non-outbreak strains were found closely related to strains from 2 outbreaks, whereas ten were genetically different from the outbreak strains. Thus, accurate tracking of HAV strains can be accomplished using HAV WG sequences, while short subgenomic regions are useful for identification of transmissions only among cases with known epidemiological association.
See on www.plosone.org