See on Scoop.it – Curiosopernatura
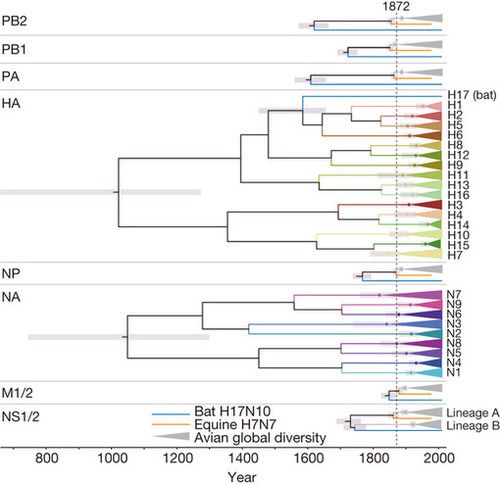
Zoonotic infectious diseases such as influenza continue to pose a grave threat to human health. However, the factors that mediate the emergence of RNA viruses such as influenza[thinsp]A virus (IAV) are still incompletely understood. Phylogenetic inference is crucial to reconstructing the origins and tracing the flow of IAV within and between hosts. Here we show that explicitly allowing IAV host lineages to have independent rates of molecular evolution is necessary for reliable phylogenetic inference of IAV and that methods that do not do so, including /`relaxed/’ molecular clock models, can be positively misleading. A phylogenomic analysis using a host-specific local clock model recovers extremely consistent evolutionary histories across all genomic segments and demonstrates that the equine H7N7 lineage is a sister clade to strains from birds[mdash]as well as those from humans, swine and the equine H3N8 lineage[mdash]sharing an ancestor with them in the mid to late 1800s. Moreover, major western and eastern hemisphere avian influenza lineages inferred for each gene coalesce in the late 1800s. On the basis of these phylogenies and the synchrony of these key nodes, we infer that the internal genes of avian influenza virus (AIV) underwent a global selective sweep beginning in the late 1800s, a process that continued throughout the twentieth century and up to the present. The resulting western hemispheric AIV lineage subsequently contributed most of the genomic segments to the 1918 pandemic virus and, independently, the 1963 equine H3N8 panzootic lineage. This approach provides a clear resolution of evolutionary patterns and processes in IAV, including the flow of viral genes and genomes within and between host lineages.
See on www.nature.com