See on Scoop.it – healthcare technology
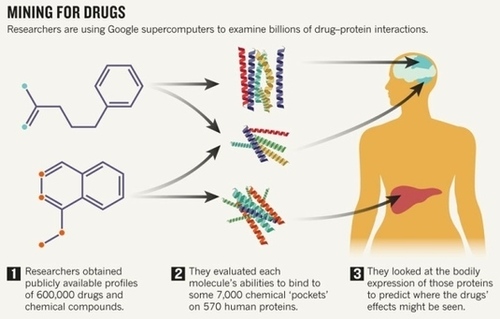
For decades, drug development was mostly a game of trial and error, with brute-force candidate screens throwing up millions more duds than winners. Researchers are now using computers to get a head start. By analysing the chemical structure of a drug, they can see if it is likely to bind to, or ‘dock’ with, a biological target such as a protein. Such algorithms are particularly useful for finding potentially toxic side effects that may come from unintended dockings to structurally similar, but untargeted, proteins.
Last week, researchers presented a computational effort that assesses billions of potential dockings on the basis of drug and protein information held in public databases. The result, a website called Drugable (drugable.com) that is backed by the US National Library of Medicine (NLM), is still in testing, but it will eventually be available for free, allowing researchers to predict how and where a compound might work in the body, purely on the basis of chemical structure
more at original : http://www.nature.com/news/project-ranks-billions-of-drug-interactions-1.14245
See on www.nature.com